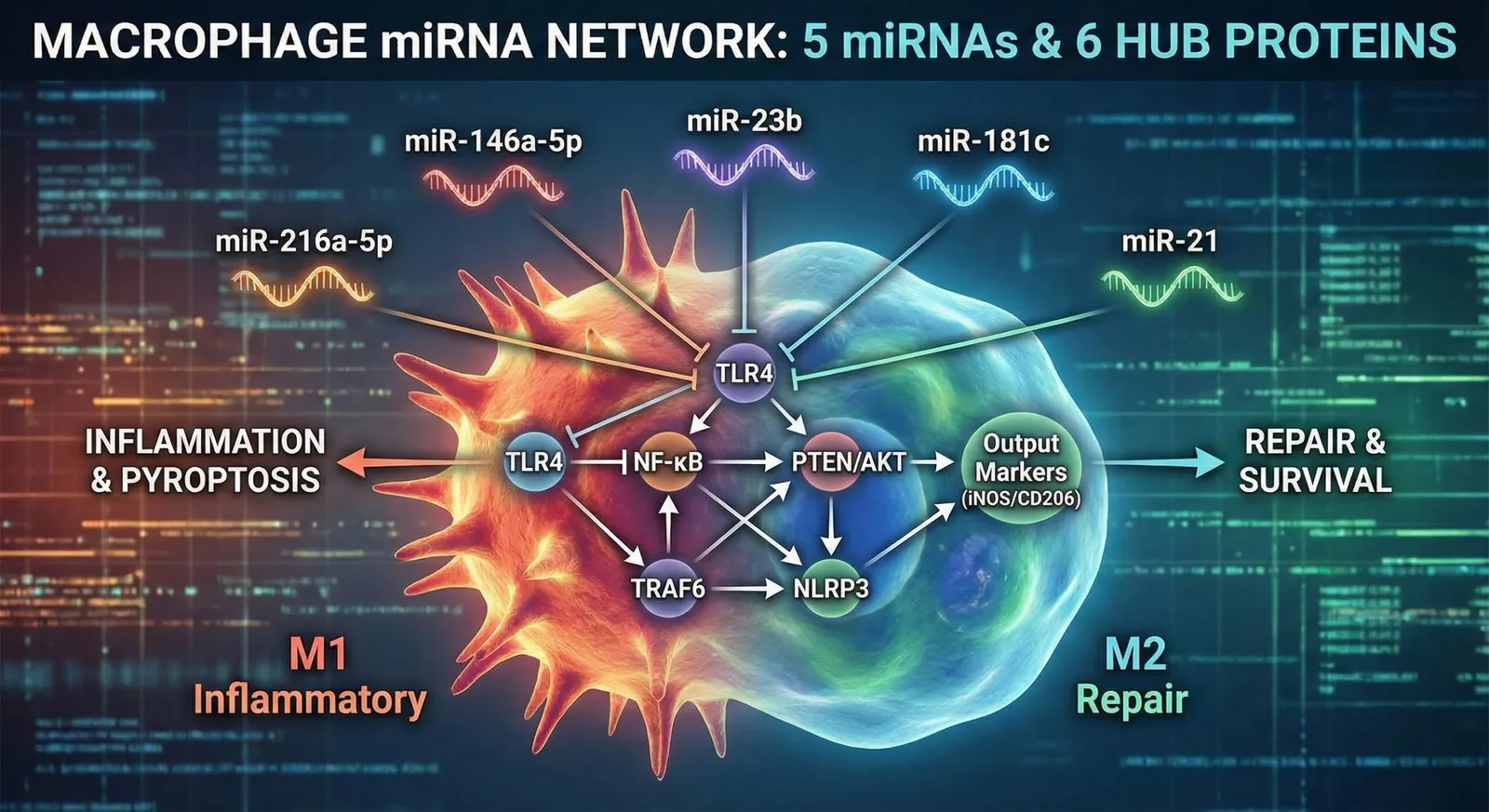
在巨噬细胞(RAW264.7、THP-1、Kupffer细胞等)的表型调控中,特定的微小RNA (miRNA) 以哪些蛋白质为靶点,并最终导向何种命运(炎症、M1/M2极化、焦亡等)。
本次,基于文献(PubMed报告),挑选出5个值得关注的微小RNA(miR‑216a‑5p / miR‑146a‑5p / miR‑23b / miR‑181c / miR‑21)。对每一个,分别整理其”5大靶蛋白”以及它们共同介入的**“信号转导的汇聚点(枢纽)”**。
1. 网络的核心:所有微小RNA共享的”6个汇聚点”
纵观这5个微小RNA所调控的蛋白质,可以看出巨噬细胞的命运决定并非分散的通路,而是汇聚于对以下主要”枢纽”的调控。通过调控这些枢纽,可诱导强有力的表型变化。
- NF‑κB(p65/p‑NF‑κB):炎症转录的最重要开关
- TLR4:炎症信号最上游的受体
- TRAF6 / IRAK1:TLR4信号的放大装置
- PTEN ↔ PI3K/AKT:生存・修复(M2)信号的拮抗轴
- NLRP3炎性小体:炎性细胞死亡(焦亡)与IL-1β成熟的执行因子
- 输出标志物群:M1(iNOS、TNF-α) vs M2(CD206、IL-10)
2. 各微小RNA的靶蛋白与表型详情
以下,针对各微小RNA,解说”已报告蛋白量/活性发生变化的5个分子”及其所引发的表型。
① miR‑216a‑5p:TLR4阻断与AKT激活的双重调控
(主要报告:Kupffer细胞、BMDM系)
这个微小RNA表现出典型的”M2诱导”动态:在切断炎症输入(TLR4)的同时,运转生存・修复(AKT)。
- TLR4 【直接靶点】:通过抑制表达来阻断下游信号。
- NF‑κB p65(磷酸化):伴随TLR4抑制,活化型减少。
- AKT(磷酸化):p-AKT升高(抗炎・生存信号的增强)。
- iNOS (NOS2):M1标志物降低。
- CD206 (MRC1):M2标志物以及Arg1升高。
表型总结 通过 TLR4↓・p‑NF‑κB↓ 以及 p‑AKT↑,在抑制炎性细胞因子的同时,强烈诱导向 M2极化(iNOS↓・CD206↑)。
② miR‑146a‑5p:炎症的”刹车角色”与NLRP3调控
(主要报告:RAW264.7、哮喘/肺模型)
作为”炎症的负调控因子”而著名,其特征在于直接打击TLR4信号的放大部分(TRAF6/IRAK1)。
- TRAF6 【直接靶点】:通过3’UTR结合抑制表达。
- IRAK1:与TRAF6一同被抑制,衰减TLR4信号。
- TLR4:在与下游因子的联动下,观察到蛋白水平的降低。
- NF‑κB(核移位/活性):活化及核内移位受到抑制。
- NLRP3:抑制炎性小体的活化,有助于M1抑制/M2促进。
表型总结 以 TRAF6/IRAK1(直接靶点) 的抑制为起点,对 NF‑κB输出与NLRP3炎性小体 进行双重抑制。展现出强有力的 抗炎(TNF-α↓, IL-10↑) 作用。
③ miR‑23b:注意因靶点而变化的”两面性”
(主要报告:THP-1、Microglia/Macrophage)
由于这个微小RNA的作用会因”以何为靶点(细胞环境)“而反转,因此在解读时需要注意。
- NF‑κB p65:有报告称经由A20抑制而活化(促炎)。
- A20 (TNFAIP3) 【直接靶点】:抑制作为NF-κB刹车的A20,从而促进炎症的系统。
- ADAM10 【直接靶点】:以此为靶点时,抑制炎症与凋亡。
- PTEN 【直接靶点】:通过抑制PTEN影响Nrf2通路与焦亡。
- NLRP3:与PTEN靶向联动,抑制焦亡(GSDMD-N, IL-1β)。
解读要点
- 促炎路线:抑制A20 → NF‑κB活化
- 抗炎/细胞保护路线:抑制ADAM10或PTEN → 抑制炎症・焦亡 ※需要判别在实验系统中哪条通路占主导。
④ miR‑181c:对TLR4/NF‑κB轴的简单抑制
(主要报告:RAW264.7)
相较于复杂的分支,主要报告描述的是直接抑制TLR4-NFκB轴。
- TLR4:通过MSC来源外泌体处理等被抑制。
- NF‑κB p65(以及p-p65):活性降低。
- TNF‑α:产生降低。
- IL‑1β:产生降低。
- IL‑10:产生升高。
表型总结 随着 TLR4/NF‑κB抑制,M1系细胞因子(TNF-α/IL-1β)降低,M2系(IL-10)升高,呈现 抗炎谱。
③ miR‑21:通过抑制PTEN实现”M2/TAM化”的关键
(主要报告:肿瘤相关巨噬细胞(TAM)、感染模型)
在肿瘤免疫的语境中频繁出现。通过抑制PTEN来运转AKT,将巨噬细胞转化为”M2样(或具免疫抑制性的TAM)”。
- PTEN:被抑制,PI3K/AKT通路的刹车被解除。
- PI3K / p‑AKT:伴随PTEN抑制而活化(信号增强)。
- STAT3:表达・活性升高(M2化的主要转录因子)。
- PDCD4:TLR4信号的放大因子。通过抑制它来负向调控NF-κB的过度活化。
- IL‑10:作为PDCD4抑制的结果,IL-10产生趋于维持・促进。
表型总结 通过 PTEN↓ → PI3K/AKT↑ → STAT3↑ 的通路,强力推进 M2/TAM化(免疫抑制)。感染时也通过PDCD4抑制而发挥过度炎症刹车的功能。
3. 整合模型:这5个微小RNA操控的”2个开关”
整合以上信息可以说,这5个微小RNA大致分为操控2个开关来改变巨噬细胞的性状。
Switch A:炎症回路的阻断(TLR4 → NF‑κB OFF)
- 负责: miR‑216a‑5p, miR‑146a‑5p, miR‑181c, miR‑21(部分)
- 机制: 通过打击TLR4、TRAF6、PDCD4等,停止NF-κB的核移位与炎性细胞因子(TNF-α, IL-1β)的产生。
- 结果: 急性炎症的平息。
Switch B:修复・生存回路的启动(PTEN → AKT ON)
- 负责: miR‑21, miR‑216a‑5p, miR‑23b(部分)
- 机制: 通过打击PTEN来 激活PI3K/AKT信号。
- 结果: M2标志物(CD206, Arg1)的升高、细胞生存、组织修复(或肿瘤促进)的转向。
4. 主要参考文献 (Key PubMed IDs)
用于本文整理的主要报告如下。详细的实验条件请确认原著。
- miR-21 / PDCD4轴 (TLR4负反馈): Sheedy et al., 2010 (PMID: 19946272)
- miR-21 / PTEN轴 (M2/TAM诱导): Lin et al., 2020 (PMID: 31814034)
- miR-146a-5p / NLRP3・M2轴 (哮喘模型): Li et al., 2022 (PMID: 35660690)
- miR-146a-5p / NLRP3轴 (NEC/THP-1): He et al., 2021 (PMID: 33585442)
- miR-23b / ADAM10轴 (抗炎): Zhang et al., 2019 (PMID: 31780861)
本博客文章基于最新的PubMed文献,整理了巨噬细胞中特定的微小RNA网络。由于行为可能因实验系统(细胞种类或刺激)而异,进行验证实验时请注意。